RNA editing represents one of the most fascinating mechanisms in molecular biology – a process that introduces targeted changes to RNA sequences after transcription, creating a dynamic transcriptome that diverges from the genome’s blueprint. Through this tutorial, you’ll learn how to analyze RNA editing events from RNA-seq data, uncovering these crucial modifications that fine-tune gene expression and cellular function.
The Hidden Language of RNA Editing
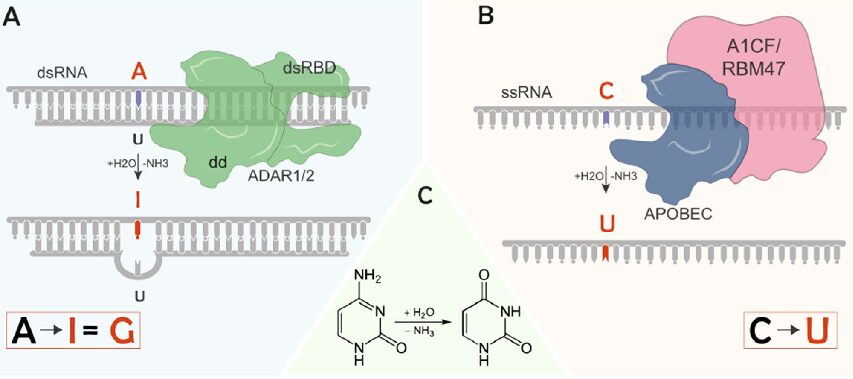
RNA editing is far more than just a molecular curiosity. When adenosine deaminases (ADARs) convert adenosine to inosine (A-to-I), or when APOBEC enzymes transform cytidine to uridine (C-to-U), they’re not just changing individual nucleotides – they’re orchestrating a complex dance of cellular regulation that impacts everything from neural function to immune response.
Think of RNA editing as nature’s sophisticated text editor, capable of:
- Rewiring neural circuits by modifying neurotransmitter receptors
- Fine-tuning immune responses through RNA sequence alterations
- Creating protein diversity without changing the genome
- Regulating gene expression through subtle molecular switches
This tutorial will guide you through the complete process of discovering and analyzing these editing events in your RNA-seq data, equipping you with both theoretical understanding and practical skills.
Choosing Your RNA Editing Analysis Tools
Before diving into the analysis, let’s explore the computational tools available for RNA editing detection. Each tool has its unique strengths, and understanding them will help you choose the right one for your research:
REDItools2: The Swiss Army Knife
This versatile suite excels at identifying A-to-I editing events with remarkable precision. Its ability to handle both RNA-only and RNA-DNA comparative analyses makes it particularly powerful for comprehensive editing studies.
GIREMI: The Statistical Pioneer
When you don’t have matched DNA sequencing data, GIREMI steps in with sophisticated statistical methods to differentiate true editing events from genetic variants. It’s particularly valuable for historical datasets or situations where DNA sequencing isn’t feasible.
RNAEditor: The User-Friendly Platform
Perfect for researchers new to RNA editing analysis, RNAEditor provides a comprehensive yet accessible interface for detection, annotation, and visualization of editing sites.
Specialized Tools for Specific Needs
- RES-Scanner2: Ideal for cross-species studies
- SPRINT: Offers streamlined analysis without SNP pre-filtering
- RESIC: Uses advanced alignment models for precise site identification
- RED-ML: Leverages machine learning for editing prediction
- DeepRed: Employs deep learning for high-sensitivity detection
Setting Up Your Analysis Environment
Let’s prepare our computational environment for RNA editing analysis using REDItools2. We’ll work in a Linux environment, using Conda for package management.
# Create a fresh conda environment
cd ~/tutorial
conda create -n Env_reditools2 python=2.7 -y
# Activate the environment
conda activate Env_reditools2
# Install essential dependencies
conda install -c conda-forge build-essential libffi libxml2 libxslt zlib bzip2 lzma ncurses openmpi mpi4py -y
conda install -c bioconda htslib samtools tabix bedtools gffread -y
# Get REDItools2 from GitHub
git clone https://github.com/tflati/reditools2.0.git
# Install REDItools2
cd reditools2.0
pip install -r requirements.txt
# Verify your installation
python src/cineca/reditools.py --help
Uncovering RNA Editing Events
Now that our environment is ready, let’s analyze our RNA-seq data for editing events. We’ll use the BAM file from our previous alignment tutorial. The reference genome for mouse can be download from refgenie as shown in the previous tutorial. Rename the downloaded genome file (fasta format) if needed. REDItools2 needs to be applied to each of your samples.
# Create an output directory
mkdir ~/tutorial/REDItools2_out
# Index your BAM file (if not already done)
samtools index SRR28119110_trimmedAligned.sortedByCoord.out.bam
# Run REDItools2 analysis
python ~/tutorial/reditools2.0/src/cineca/reditools.py -S \
-f SRR28119110_trimmedAligned.sortedByCoord.out.bam \
-r /your_reference_genome_directory/GRCm38.genome.fa \
-o ~/tutorial/REDItools2_out/REDITools2_Outputs_SRR28119110.tsv

Understanding Your Results
REDItools2 generates a detailed output file (above) containing crucial information about each potential editing site. Here’s what each column tells you:
Location Information:
- Region: Chromosome identifier (e.g., chr1, chrX)
- Position: Precise genomic coordinate
- Reference: Original nucleotide in the genome
- Strand: DNA strand (+/-) location
Quality Metrics:
- Coverage-q30: High-quality read count
- MeanQ: Average base quality score
- BaseCount: Distribution of A, C, G, T at the site
Editing Details:
- AllSubs: Observed nucleotide changes
- Frequency: Editing ratio calculation
Advanced Analysis: Differential Editing
One of the most challenging aspects of RNA editing analysis is comparing editing patterns between conditions. Here’s why it’s complex:
- Sample Variability: Editing sites often show high individual variation
- Coverage Issues: Not all sites are equally covered across samples
- Technical Challenges: Low-frequency editing events can be hard to detect consistently
Let’s tackle these challenges with a systematic R-based analysis. Essential R packages can be installed as shown in the previous tutorial. I understand the R code ahead might look daunting if you’re new to programming. Don’t worry – while we’ll explore R programming in detail in a future tutorial series, for now let’s focus on understanding what each block of code accomplishes. Think of it as reading a recipe before learning to cook – you can grasp the overall process even if you’re not familiar with all the techniques yet.
Let’s break down what this code does step by step:
- First, it performs quality control by filtering out unreliable editing events that don’t meet our quality thresholds
- Then, it removes editing sites that only appear in one sample (we need sites present across samples for comparison)
- Next, it identifies and removes unchanging sites – those with identical frequencies across all samples (they won’t be informative for our differential analysis)
- Finally, it combines the editing frequencies from all four samples into a single unified table (redi_dt_merge)
To prepare this data for statistical analysis, we normalize the editing frequencies using a mathematical transformation called Arcsine Square Root. This transformation helps ensure our data meets the statistical assumptions needed for differential analysis.
This organized workflow takes us from raw editing data to a clean, analysis-ready dataset. As you become more comfortable with R, you’ll find these data processing steps become more intuitive.
# Load essential packages
library(data.table)
library(limma)
library(edgeR)
# Import REDItools2 results
redi_dt <- lapply(list.files("~/tutorial/REDItools2_out",
"REDITools2_Outputs_.*.tsv",
full.names = T),
fread,
data.table = F)
# Name your samples
names(redi_dt) <- gsub("REDITools2_Outputs_|\\.tsv", "",
list.files("~/tutorial/REDItools2_out",
"REDITools2_Outputs_.*.tsv"))
# Filter and process editing sites
redi_dt <- lapply(redi_dt, \(x) {
x[x$`Coverage-q30` >= 10 & x$Frequency >= 0.01, ]
})
# Create unique identifiers
redi_dt <- lapply(redi_dt, \(x) {
x$EventID <- paste(x$Region, x$Position, x$AllSubs, sep = "_")
return(x[, c("EventID", "Frequency")])
})
# Rename the frequency column of each table
redi_dt <- mapply(\(x, y) {colnames(x)[2] <- y; return(x)},
redi_dt, names(redi_dt), SIMPLIFY = F)
# Merge editing results
redi_dt_merge <- Reduce(\(x, y) merge(x, y, by = "EventID", all = TRUE), redi_dt)
redi_dt_merge <- na.omit(redi_dt_merge)
rownames(redi_dt_merge) <- redi_dt_merge$EventID
redi_dt_merge <- redi_dt_merge[, -1]
# Remove unchanging editing sites
redi_dt_merge <- redi_dt_merge[apply(redi_dt_merge, 1, \(x) length(unique(x)) > 1), ]
# Normalize the editing frequencies
redi_dt_merge <- apply(redi_dt_merge, 2, function(col) asin(sqrt(col)))
# Make sample information table
meta <- data.frame(SampleID = colnames(redi_dt_merge),
Treatment = c("KRAS_SPIB", "KRAS_SPIB", "KRAS", "KRAS"))
rownames(meta) <- meta$SampleID
# Make DGEList by combing sample information and the editing frequencies
dge <- DGEList(counts=redi_dt_merge, samples = meta)
Statistical Analysis of Differential Editing
While differential editing analysis shares some statistical principles with differential gene expression analysis, we’ll now adapt those methods to analyze editing frequencies rather than gene counts. Though we’re using similar statistical tools from the limma package, we’re applying them in a more specialized way to identify significant changes in RNA editing patterns between conditions.
# Set up your experimental design
design <- model.matrix(~ 0 + dge$samples[["Treatment"]])
colnames(design) <- gsub(".*]]", "", colnames(design))
# Create contrast matrix
contrast_matrix <- makeContrasts(contrasts = comparison,
levels = design)
# Fit statistical model
fit <- lmFit(dge$counts, design)
fit_2 <- contrasts.fit(fit, contrast_matrix)
fit_2 <- eBayes(fit_2)
# Extract results
deg_tbl <- topTable(fit_2,
coef = colnames(contrast_matrix),
n = Inf,
p.value=1,
lfc=0,
sort.by = "p")
# Save the results
fwrite(deg_tbl, "Diff_Editing_P1_lfc0.tsv", sep = "\t", row.names = T)
# Make a bed file for the differential results for annotation purpose
deg_bed <- data.frame(sapply(strsplit(rownames(deg_tbl), "_"), "[[", 1), sapply(strsplit(rownames(deg_tbl), "_"), "[[", 2), sapply(strsplit(rownames(deg_tbl), "_"), "[[", 2))
fwrite(deg_bed, "Diff_Editing_P1_lfc0.bed", sep = "\t", col.names = F)

Annotating the Differential Editing Sites
By mapping our identified differential editing sites to genomic annotations, we determined which genes contain or are associated with these sites.
# Convert genomic annotation file (GTF) to bed format
gffread gencode.vM25.annotation.gtf -T -o gencode.vM25.annotation.bed
# Annotate differential editing sites
bedtools intersect -a Diff_Editing_P1_lfc0.bed -b gencode.vM25.annotation.bed -wa -wb > annotated_Diff_Editing_P1_lfc0.bed

Conclusion
RNA editing analysis reveals a crucial layer of transcriptome regulation that goes beyond simple gene expression. Through careful analysis of these modifications, we can better understand how cells fine-tune their molecular functions and respond to changing conditions.
As you continue your journey in RNA-seq analysis, remember that RNA editing is just one piece of the complex puzzle of gene regulation. Each editing site you discover could be a key to understanding cellular adaptation and response.
References
- Morales, D.R.; Rennie, S.; Uchida, S. Benchmarking RNA Editing Detection Tools. BioTech 2023, 12, 56. https://doi.org/10.3390/biotech12030056
- Birgaoanu M, Sachse M, Gatsiou A. RNA Editing Therapeutics: Advances, Challenges and Perspectives on Combating Heart Disease. Cardiovasc Drugs Ther. 2023 Apr;37(2):401-411. doi: 10.1007/s10557-022-07391-3. Epub 2022 Oct 14. PMID: 36239832; PMCID: PMC9561330.




Leave a Reply