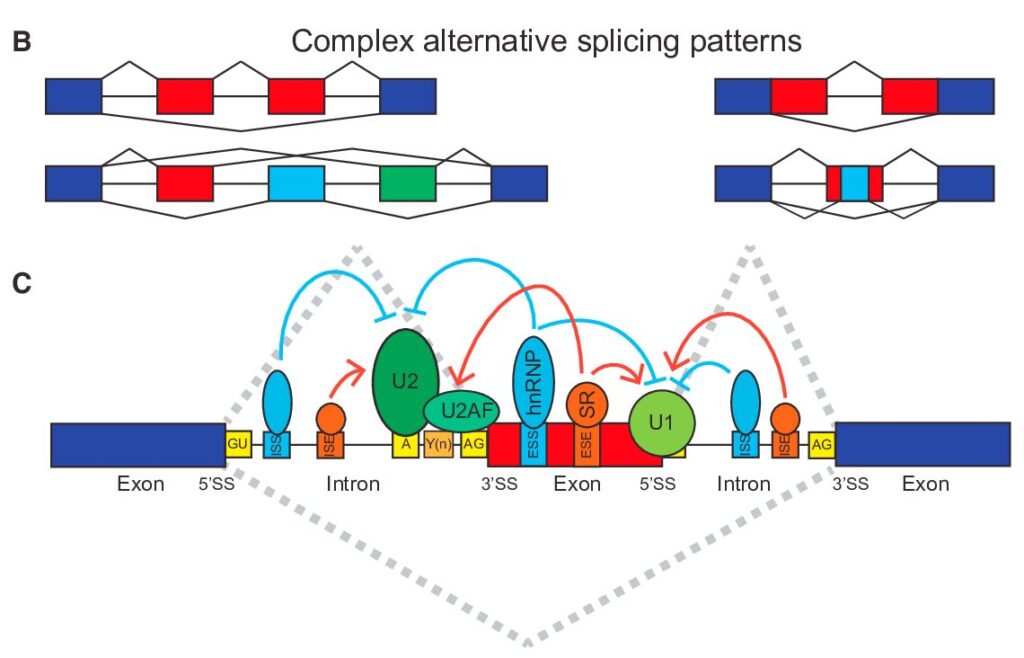
From Gene-Level to Transcript-Level Analysis
In our previous exploration of gene-level splicing analysis, we laid the groundwork for understanding how alternative splicing shapes gene expression. Now, we’re taking a deeper dive into the fascinating world of transcript-level analysis, where we can uncover the intricate details of how genes produce different protein variants through alternative splicing.
Think of gene-level analysis as viewing a city from a satellite – you see the overall structure and major changes. Transcript-level analysis, however, is like walking through the streets themselves, examining each building and understanding how they contribute to the city’s function. This granular perspective reveals crucial details that might otherwise be missed.
Understanding the Power of Transcript-Level Analysis
While gene-level analysis excels at identifying broad splicing patterns, transcript-level analysis reveals the sophisticated mechanisms that create protein diversity. This approach becomes particularly powerful when:
- Investigating complex diseases where specific protein variants play crucial roles
- Developing targeted therapies that need to address particular transcript isoforms
- Studying developmental processes where precise protein variants are required
- Understanding evolutionary adaptations through transcript diversity
Real-World Impact in Medical Research
The applications of transcript-level splicing analysis in medicine have been transformative. Consider spinal muscular atrophy (SMA), where understanding specific SMN2 transcript variants led to the development of Spinraza, a life-changing therapy. Similar successes are emerging in:
- Cancer treatment: Identifying isoform-specific biomarkers for more accurate diagnosis
- Neurodegenerative diseases: Understanding how aberrant splicing contributes to pathology
- Personalized medicine: Tailoring treatments based on patient-specific splicing patterns
- Drug development: Creating therapies that target specific transcript variants
SUPPA2: A Powerful Tool for Splicing Analysis
SUPPA2 has emerged as a leading tool for transcript-level splicing analysis, combining efficiency with comprehensive functionality. Its ability to process large datasets while maintaining accuracy makes it particularly valuable for modern genomic research.
Getting Started with SUPPA2
Let’s set up our analysis environment. The following commands create a clean workspace for our analysis:
# Create a fresh conda environment for SUPPA2
conda create -n suppa2_env python=3.8 -y
# Activate our new environment
conda activate suppa2_env
# Download SUPPA2 from the official repository
git clone https://github.com/comprna/SUPPA.git
The Analysis Pipeline: A Step-by-Step Journey
Step 1: Creating the Foundation – Generating IOE Files
Our first task is to generate IOE files, which map the relationships between splicing events and transcripts. Make sure you specify the correct paths of the tools and files on your system.
# Generate comprehensive IOE files for all splicing types
python ~/SUPPA/suppa.py generateEvents \
-i ~/Genome_Index/GTF/GRCm38/gencode.vM25.annotation.gtf \
-o IOE \
-f ioe \
-e SE SS MX RI FL
# This command creates both GTF and IOE files for:
# - Skipped exons (SE)
# - Alternative splice sites (A5/A3)
# - Mutually exclusive exons (MX)
# - Retained introns (RI)
# - Alternative first/last exons (AF/AL)
Step 2: Quantifying Splicing Events – PSI Calculation
Next, we’ll calculate PSI (Percent Spliced In) values, which tell us the relative abundance of each splicing event. This requires the IOE files generated in Step 1 and the transcript/isoform expression values (TPM). For this tutorial, we use the TPM data from our previous isoform analysis tutorial, which are stored in the quant.sf files.
Here, we quantify PSI values for two treatment groups: “KRAS” and “KRAS_SPIB.” The required format of the TPM matrix for each group is shown below. Note that the first column must only contain sample names. The files should be tab-delimited (TSV format). You can create TSV files by saving a spreadsheet as “Tab Delimited Text (.txt)” in Microsoft Excel.

The process of calculating PSI is demonstrated below. The results will be saved in the output files specified by the -o option in the commands. These PSI files will contain the relative abundance for each splicing event in each treatment group.

# Calculate PSI values for A3 events in both conditions
python ~/SUPPA/suppa.py psiPerEvent \
-i IOE_A3_strict.ioe \
-e KRAS_SPIB_tmp.tsv \
-o PSI_KRAS_SPIB_A3
python ~/SUPPA/suppa.py psiPerEvent \
-i IOE_A3_strict.ioe \
-e KRAS_tmp.tsv \
-o PSI_KRAS_A3
# Similar commands are used for other splicing events (A5, AF, AL, MX, RI, SE)
PSI_KRAS_SPIB_A3.psi

PSI_KRAS_A3.psi

Step 3: Identifying Significant Changes – Differential Analysis
Finally, we’ll identify significant splicing changes between conditions.
The differential splicing analysis requires the PSI files generated in Step 2 and the TMP (transcript expression) files as input.
If your dataset includes more than two treatment groups, you can incorporate them into the commands using the format demonstrated below. The -c option will automatically compare all pairwise combinations among the groups.
# Perform differential splicing analysis for A3 events
python ~/SUPPA/suppa.py diffSplice \
-m empirical \
-i IOE_A3_strict.ioe \
-p PSI_KRAS_SPIB_A3.psi PSI_KRAS_A3.psi \
-e KRAS_SPIB_tmp.tsv KRAS_tmp.tsv \
-gc -c \
-o diffSplice_A3
# Similar commands are used for other splicing events (A5, AF, AL, MX, RI, SE)
The differential splicing analysis produces a dpsi file, containing PSI differences and p-values, and a psivec file, which includes the PSI values for each sample for each splicing event.
The dpsi file:

The psivec file:

The results of the differential splicing analysis can be filtered (e.g., by p-value < 0.05), combined, summarized, and visualized using Microsoft Excel or R, depending on your familiarity with the tools. The pie chart below provides a summary of the differential splicing analysis described above.

Conclusion
Transcript-level alternative splicing analysis represents a powerful approach for understanding gene regulation and disease mechanisms. Through SUPPA2 and the workflow described here, researchers can uncover crucial insights into splicing patterns and their biological implications. As sequencing technologies and analysis tools continue to evolve, the ability to accurately quantify and interpret splicing events becomes increasingly valuable for both basic research and clinical applications. By following this guide, you’ll be well-equipped to conduct thorough and reliable splicing analysis that can contribute to our understanding of gene regulation and disease mechanisms.
References
- Trincado, J.L., et al. (2018). SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biology, 19, 40. https://doi.org/10.1186/s13059-018-1417-1
- Park E, Pan Z, Zhang Z, Lin L, Xing Y. The Expanding Landscape of Alternative Splicing Variation in Human Populations. Am J Hum Genet. 2018 Jan 4;102(1):11-26. doi: 10.1016/j.ajhg.2017.11.002. PMID: 29304370; PMCID: PMC5777382.




Leave a Reply